
Press release
FDA Approves RECOVER IV Randomized Controlled Trial with Exception from Informed Consent (EFIC)
FDA Also Approves and Closes RECOVER III Post-Approval Study
DANVERS, Mass. – September 16, 2022 – Abiomed (Nasdaq: ABMD) announces two approvals from the U.S. Food and Drug Administration (FDA) related to clinical research of Impella® heart pumps in acute myocardial infarction (AMI) cardiogenic shock patients.
The FDA has approved the on-label RECOVER IV randomized controlled trial (RCT) for AMI cardiogenic shock patients. RECOVER IV is a two-arm trial that will assess whether percutaneous coronary intervention (PCI), with Impella support initiated prior to the PCI, is superior to PCI without Impella support.
“This landmark trial will be the culmination of over 20 years of research in the interventional therapy of AMI and will apply all the clinical advancements we have made to improve survival and heart recovery for AMI patients with cardiogenic shock as demonstrated in multiple prospective studies,” said William O’Neill, MD, medical director of the Center for Structural Heart Disease at Henry Ford Health and a RECOVER IV national co-principal investigator.
The primary endpoint of RECOVER IV is all-cause mortality at 30 days. Secondary endpoints include major adverse cardiovascular and cerebrovascular events (MACCE) at 30 days, days alive out of the hospital at six months, recovery of left ventricular (LV) function, need for durable ventricular assist device (VAD) or heart transplant, and health-related quality of life as measured by responses to the Kansas City Cardiomyopathy Questionnaire (KCCQ) at one year. Abiomed’s goal in conducting the trial is to achieve a global AMI cardiogenic shock Class I guideline recommendation for Impella and related best practice protocols, including Impella implantation pre-PCI (see figure 1).
“I am optimistic that RECOVER IV will further demonstrate the benefits of hemodynamic support and best practice protocols. These benefits include ventricular unloading using Impella pre-PCI, reduced LV wall stress, reduced pulmonary congestion, increased collateral coronary blood flow, and enhanced cardio protection so that more AMI cardiogenic shock patients can survive and achieve native heart recovery. The heart team and field have evolved and understand how important myocardial recovery is for both AMI and AMI cardiogenic shock to reduce the growing epidemic of heart failure,” said Navin K. Kapur, MD, executive director of the Cardiovascular Center for Research and Innovation (CVCRI) at Tufts Medical Center and a national co-principal investigator for RECOVER IV.
FDA Approves and Closes RECOVER III Post-Approval Study
Additionally, the FDA has approved and closed Impella’s prospective AMI cardiogenic shock post-approval study (PAS), RECOVER III. This study gathered real world evidence on AMI cardiogenic shock patients treated with Impella between 2017 – 2019, collecting detailed data including stages of cardiogenic shock, cardiac output, and timing of implantation. RECOVER III fulfills Abiomed’s PAS requirement and the FDA’s approval and closure of RECOVER III further validates Impella as a safe and effective therapy for AMI cardiogenic shock.
Impella remains the only mechanical circulatory support (MCS) device that has received the FDA’s highest level of pre-market approval (PMA) and PAS regulatory approval for AMI cardiogenic shock. Based on RECOVER III, Impella’s label for AMI cardiogenic shock will be updated to reflect data for up to one-year post-procedure.
AMI Cardiogenic Shock Clinical History
AMI cardiogenic shock has one of the highest mortality rates in the field of medicine. The survival rate has remained approximately 50% for cardiogenic shock patients in SCAI stages D and E without Impella support and associated best practices. Survival alone in cardiogenic shock is no longer the gold standard. Multiple Impella best practice studies demonstrate greater than 70% survival with greater than 90% native heart recovery (see figure 2). Impella best practices, such as pre-PCI implantation (see figure 3), have been developed by the recognized physician experts in the field of circulatory support and published over the last decade in multiple clinical studies from the U.S., Germany, Italy and Japan. Heart recovery after AMI cardiogenic shock improves patient quality of life and makes Impella one of the most cost-effective therapies in the CMS Medicare population and in private insurance. In the U.S. alone, more than 200,000 patients are admitted to the hospital every year in cardiogenic shock.
FDA Regulatory History of AMI Cardiogenic Shock
Impella is the most studied heart pump in the history of the FDA (see figures 4 & 5) and has exclusive FDA PMA as a safe and effective therapy for cardiogenic shock, high-risk PCI and right heart failure. Since 2004, more than 1,200 peer-reviewed studies, including real-world evidence analyses, prospective clinical studies and RCTs have published about the clinical benefits of Impella.
Impella has been used to treat more than 235,000 patients globally and is included in 13 clinical society guidelines. In 2021, the European Society of Cardiology upgraded Impella to a Class IIa recommendation for the treatment of cardiogenic shock. The intra-aortic balloon (IAB) is currently Class III (harmful) for routine use in cardiogenic shock in Europe and Japan based on the IABP-SHOCK II RCT, which demonstrated IAB compared to inotropic therapy provided no benefit to survival or hemodynamic augmentation. In 2020, IAB became Class III (harmful) in the U.S. guideline recommendations for post-cardiotomy cardiogenic shock.
All MCS and ventricular assist devices (VADs) since 1992 were approved with single arm studies compared to historical survival rates judged on objective performance criteria (OPC) due to the ethical and logistical challenges of randomizing critically ill patients requiring immediate hemodynamic augmentation. In 2008 and 2009, Abiomed attempted the FDA RECOVER II RCT, which compared Impella to IAB in AMI cardiogenic shock. RECOVER II enrolled only 1 patient in 15 months at more than 30 sites before being halted for logistical and ethical consent challenges and lack of enrollment.
The FDA granted Impella 510(k) clearance in 2008 and after multiple FDA and prospective physician-initiated studies, the FDA approved high-risk PCI in 2015, AMI cardiogenic shock in 2016 (see figure 6), and other forms of heart failure with cardiogenic shock in 2018. With the fulfillment of RECOVER III and approval of RECOVER IV RCT, Abiomed is pursuing an on-label study to strengthen the global guidelines and improve outcomes for patients.
Abiomed has sponsored and funded several AMI cardiogenic shock studies since 2006 (see figure 7) including the only FDA studies in this space. The difficulty randomizing AMI cardiogenic shock patients in execution has been demonstrated in multiple studies including IMPRESS in STEMI (n=18), IMPRESS in Cardiac Arrest (n=48), Seyfarth, et al. (n=26) and the Abiomed-sponsored FDA RECOVER II RCT (n=1). All these studies failed to randomize and were halted early for failure to enroll their designated numbers based on logistical and ethical consent challenges.
Recent FDA Regulatory Changes for Exception from Informed Consent (EFIC):
Randomizing patients in AMICS has been challenging because they require emergent care and are too sick to provide traditional informed consent to enroll in a trial. In 1996, the FDA created the exception from informed consent (EFIC) pathway for emergency clinical research. This pathway allows investigators to broadly educate a community about a trial, then enroll patients without consent from patients, their family or their legally authorized representatives.
In 2022, after engaging with the FDA on the RECOVER IV RCT study design, the FDA approved the RECOVER IV RCT study protocol, which includes the use of EFIC. This community awareness process is rare and only used when the patients being studied are experiencing a life-threatening medical condition causing serious deficiency of mental function. This is a significant milestone for the field and physician leaders. The next steps include local hospital Institutional Review Board (IRB) approvals and a commitment from physicians to randomize.
“This pivotal randomized trial is historic as the first to use EFIC community consent to enroll cardiogenic shock patients. I applaud the FDA for its partnership to help solve the consent challenges in cardiogenic shock RCTs and call on the physician community to enroll and randomize patients in RECOVER IV,” said Gregg W. Stone, MD, professor of medicine and director of academic affairs for the Mount Sinai Heart Health System in New York and study chair for RECOVER IV.
For more information on current best practices in treating AMI cardiogenic shock patients please click here.
For more information on the RECOVER IV RCT study please click here.
Figure 1.
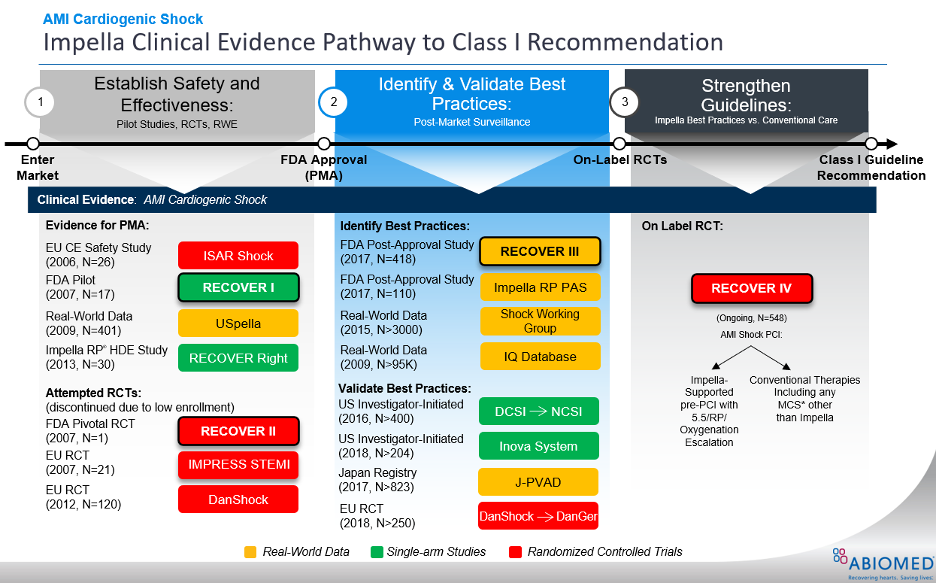
RECOVER IV is an on-label, two arm RCT that is designed to provide the clinical evidence needed to achieve a Class I guideline recommendation for Impella use in AMI cardiogenic shock.
Figure 2.
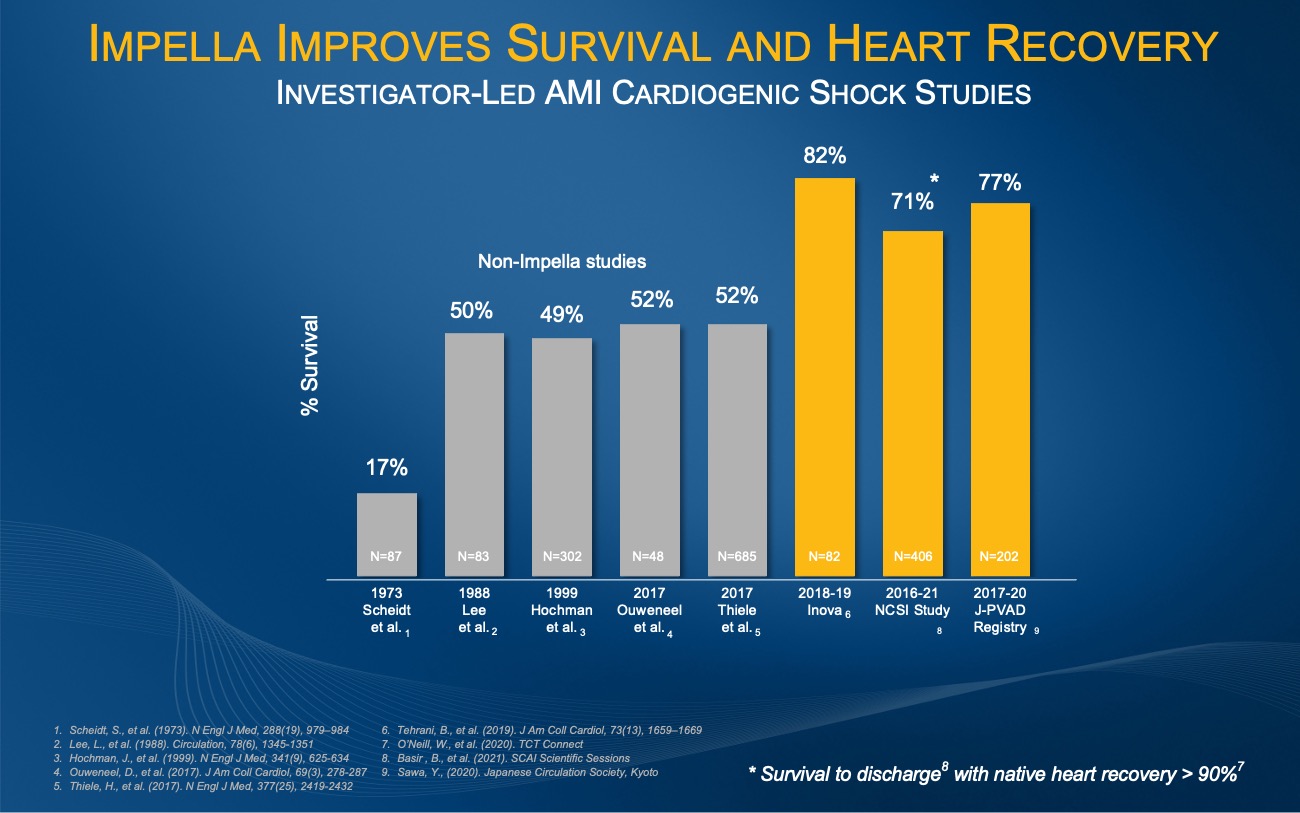
The investigator-led Inova, NCSI and J-PVAD studies all demonstrate an improvement from the historical AMI cardiogenic shock survival rate of approximately 50% when patients are treated with best practices including Impella.
Figure 3.
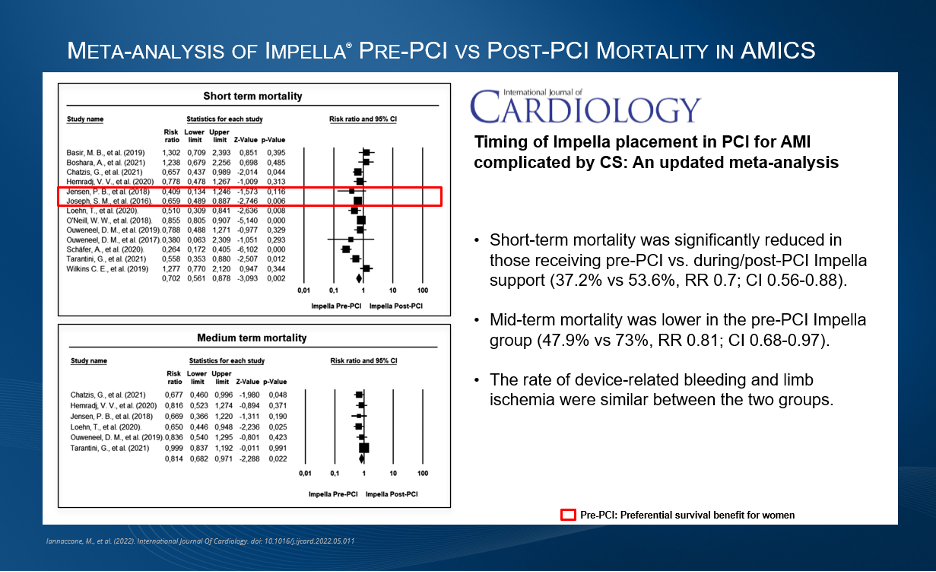
A 2022 meta-analysis demonstrated a reduction in short-term mortality and lower mid-term mortality for patients receiving Impella pre-PCI vs. post-PCI in AMICS.
Figure 4.
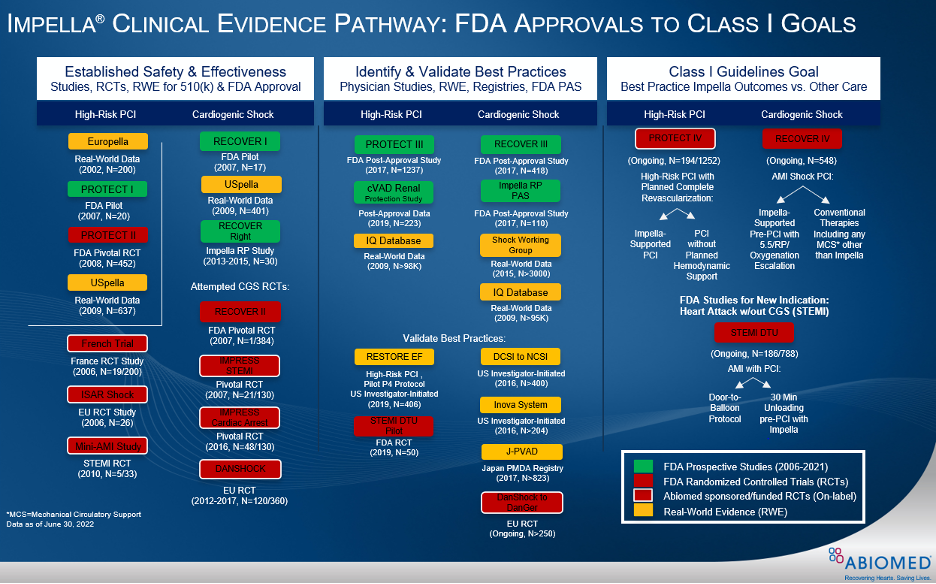
Impella is the most studied MCS device in the history of the FDA. Since 2004, more than 1,200 peer-reviewed studies, including real-world evidence analyses, prospective clinical studies and RCTs have published about the clinical benefits and cost effectiveness of Impella.
Figure 5.

Impella remains the most studied heart pump in the history of the FDA from 2006-2022, fueled by rigorous levels of evidence and high-quality data sources.
Figure 6.
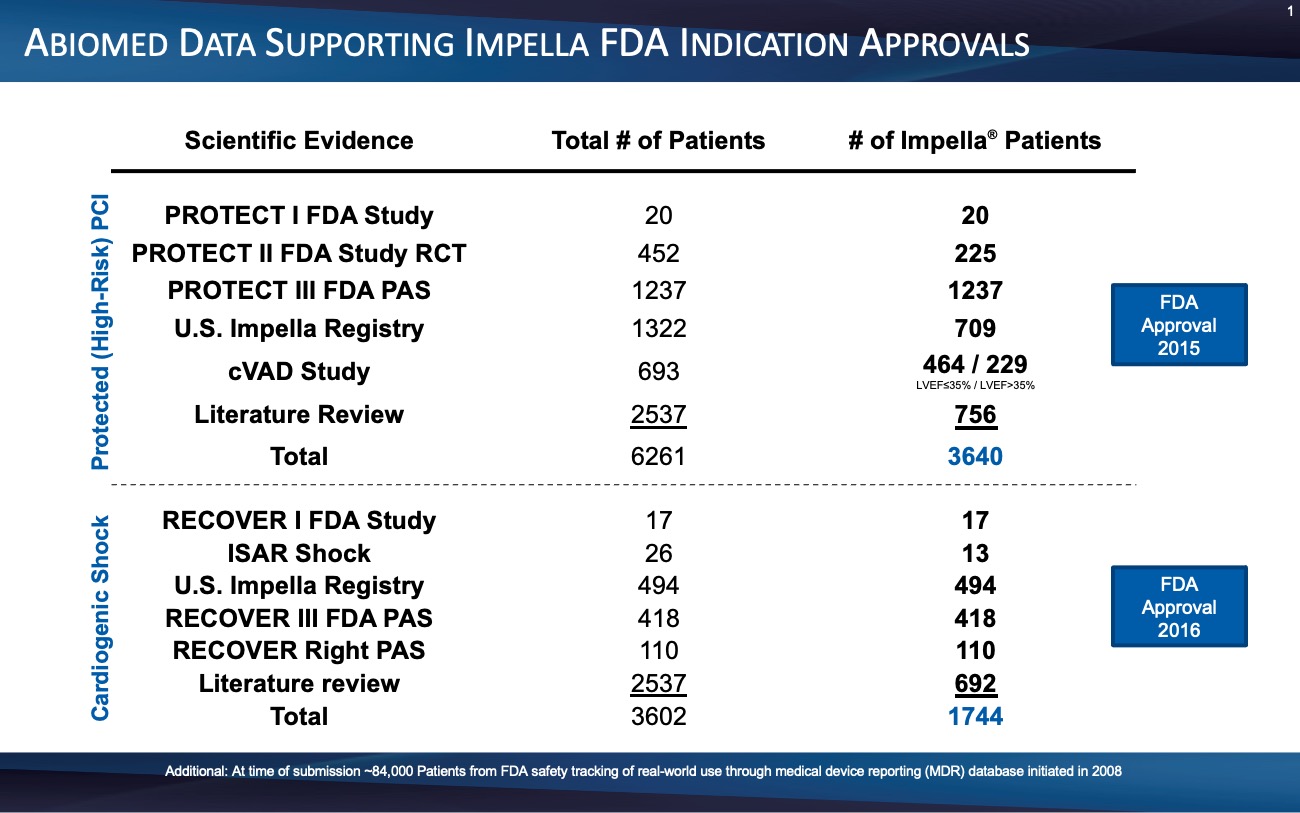
After multiple FDA and prospective physician-initiated studies, the FDA granted Impella a PMA for AMI cardiogenic shock in 2016.
Figure 7.
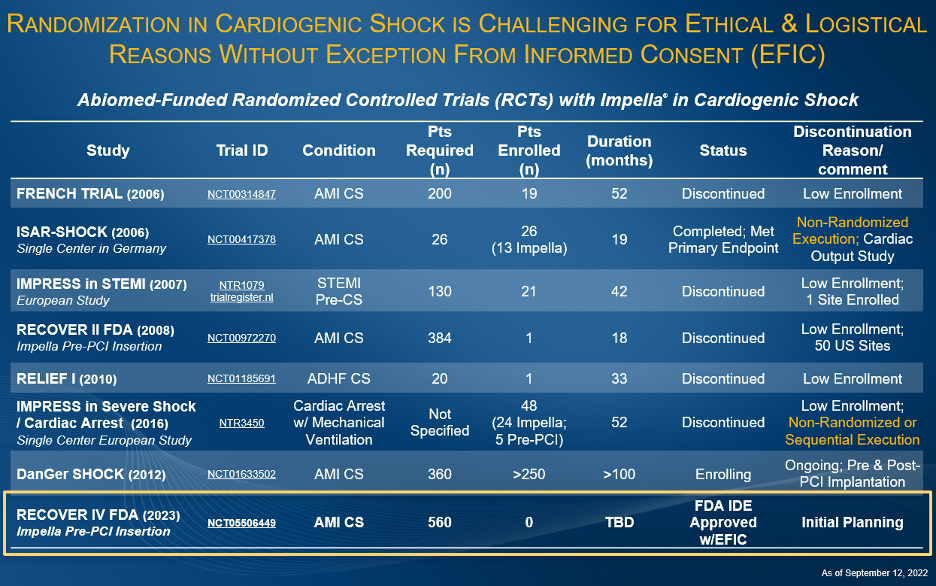
Abiomed has sponsored and funded several AMI cardiogenic shock studies since 2006 including the only FDA studies. Difficulty in randomization has been demonstrated in multiple studies including IMPRESS in STEMI (n=18), IMPRESS in Cardiac Arrest (n=48), Seyfarth, et al. (n=26) and the Abiomed-sponsored FDA RECOVER II RCT (n=1). All these studies failed to randomize in execution and were halted early for failure to enroll their designated numbers.
ABOUT IMPELLA HEART PUMPS
Impella 2.5® and Impella CP® with SmartAssist® are U.S. FDA approved to treat certain advanced heart failure patients undergoing elective and urgent percutaneous coronary interventions (PCI), such as stenting or balloon angioplasty, to reopen blocked coronary arteries.
Impella 2.5, Impella CP, Impella CP with SmartAssist®, Impella 5.0®, Impella LD®, and Impella 5.5® with SmartAssist® are U.S. FDA approved to treat heart attack or cardiomyopathy patients in cardiogenic shock and have the unique ability to enable native heart recovery, allowing patients to return home with their own heart.
ABOUT ABIOMED
Based in Danvers, Massachusetts, U.S.A., Abiomed is a leading provider of medical technology that provides circulatory support and oxygenation. Our products are designed to enable the heart to rest by improving blood flow and/or provide sufficient oxygenation to those in respiratory failure. For additional information, please visit: http://www.abiomed.com/.
FORWARD-LOOKING STATEMENTS
Any forward-looking statements are subject to risks and uncertainties such as those described in Abiomed's periodic reports on file with the Securities and Exchange Commission. Actual results may differ materially from anticipated results.
COR-0727